![]()
Copyright Ross Walker 2006
TUTORIAL B4
Using Antechamber to Create Leap Input Files for Simulating Sustiva using
the General Amber Force Field
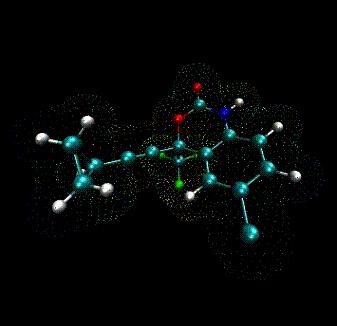
By Ross Walker
訳
大阪府立大学・生命環境科学研究科
応用生命科学専攻・生体情報化学研究室
和田野 晃
このチュートリアルでは、Leapで使用できる有機化合物に対するprmtop と inpcrd ファイルを作成しAMBER 8でシミュレーションが出来るようにすることを目的とする。そのために、同梱されているAntechamber toolsのマニュアルを記述する。
Antechamber は"general AMBER force field (GAFF)"とともに使用する。このforce field は特にほとんどのpharmaceutical
molecules をカバーする様に作成され、traditional AMBER force fields と互換性があり、シミュレーションでは一緒に用いることが可能である。traditional
AMBER force fieldsと同様に, GAFFは結合と結合角にたいしてa simple harmonic function formを用いるが、 一般のタンパク質 と DNA に対応した AMBER force fields とは異なりGAFF で使用されている原子種はより一般的で、ほとんどの有機化合物に含まれる原子をカバーしている。現在、GAFF force field は33 basic atom types と 22 special atom typesを含んでいる。 charge methods としては HF/6-31G* RESP or AM1-BCC2が使用可能である。
目的的に, GAFFは完全なforce field で、パラメータが存在しない場合はまれであり、有機化学で現れるC, N, O, S, P, H, F, Cl, Br および Iをカバーしている。さらに、wince GAFF は、AMBER macromolecular force
fieldsと完全に互換性があり、合理的ドラッグデザインに対する分子動力学的ツールになるであろう。特に、結合自由エネルギーの計算や分子ドッキング研究では重要になると思われる。
Antechamber tool set は、AMBER simulation programs に使用するtopology
files を短時間で作れるように設計されている。以前のtutorialでは、マニュアルで原子タイプをアサインしていたが、 antechamber はGAFF により自動的にアサインすることを可能にしている。Antechamberは以下の問題を解決する。
1. Automatically
identify bond and atom types
2. Judge
atomic equivalence
3. Generate
residue topology files
4. Find
missing force field parameters and supply reasonable suggestions
しかし、Antechamber は必ずしも完全に上記の問題を解決するわけではない。従って、常にAntechamberがアサインした原子タイプを自分で注意深くチェックする必要がある。Scientific softwareを内容がわからないまま、"Black Box" として使用しないようにしましょう!
------------------------------------------------------------------------
TUTORIAL B4 - SECTION 2
Creating topology and coordinate files for Sustiva
このチュートリアルでは、Antechamber
toolsをLeap と共に用い、医薬品Sustiva
(efavirenz)のtopology と coordinate files を作成します。 Sustiva (http://www.sustiva.com) はHIV-1specific,
non-nucleoside, reverse transcriptase inhibitor で Bristol Myers Squibb により販売されており、人間に感染したHIVの進行をコントロールするために使われている。正式名は (S)-6-chloro-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-oneで構造式はC14H9ClF3NO2
、2D構造は下記に示す。
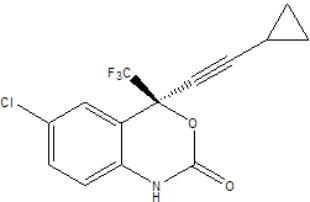
sustiva.pdb
なにか、PDBファイルを表示できるソフトでこの構造を、確認してみてください。
Antechamberで この分子の原子タイプをアサインし、a set of
point charges を計算します。AntechamberはAntechamber toolsセットのなかで一番重要なソフトである。 このソフトは、多くのファイルの変換と原子のチャージとタイプをアサインする。Antechamberは必要につれ、以下のプログラムを実行するがそれら全てamber8に含まれている。: divcon, atomtype, am1bcc, bondtype, espgen, respgen と prepgenである。 さらに大文字で表記される中間ファイルを多く作成する。
早速、antechamber を sustiva pdb fileに適応してみる。
"prepin" file を作成するために、まず下記のcommandを使う。
|
>antechamber -i sustiva.pdb -fi pdb -o
sustiva.prepin -fo prepi -c bcc -s 2 |
ここで「-i sustiva.pdb」は3D structure fileを指定し、「 -fi pdb」は入力ファイルがpdbファイルであることを示しています。 (これ以外にも、Gaussian
Z-Matrix [gzmat], Gaussian Output [gout], MDL [mdl], amber Restart [rst], Sybyl
Mol2 [mol2]が使えます). 「 -o
sustiva.prepin」は出力ファイル名を指定し、「-fo prepi 」はそのタイプがamber PREP formatで有ることを指定しています。 (このフォーマットはLeapで使えるものです). 「 -c bcc」オプションはantechamberがBCC charge model を使用してatomic
point charges計算し、「-s 2」オプションは antechamber により作られるstatus information がverbosity(冗長性)であることを意味しています(ちょっと内容を理解不能?)。今回のケースでは2を選択している。以上を踏まえて上記コマンドを実行すると、画面の表示は表1のようになる。
表1 antechamberの実行
|
Running: /usr/local/amber8/exe/bondtype -i
ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac -j full Running: /usr/local/amber8/exe/atomtype -i
ANTECHAMBER_AC.AC0 -o ANTECHAMBER_AC.AC -p gaff Total number of electrons: 160; net charge:
0 Running: /usr/local/amber8/exe/divcon Running: /usr/local/amber8/exe/am1bcc -i
ANTECHAMBER_AM1BCC_PRE.AC -o ANTECHAMBER_AM1BCC.AC -f ac -p /usr/local/amber8/dat/antechamber/BCCPARM.DAT
-s 2 -j 1 Running: /usr/local/amber8/exe/atomtype -f
ac -p bcc -o ANTECHAMBER_AM1BCC.AC -i ANTECHAMBER_AM1BCC_PRE.AC Running: /usr/local/amber8/exe/atomtype -i
ANTECHAMBER_PREP.AC0 -o ANTECHAMBER_PREP.AC -p gaff Running: /usr/local/amber8/exe/prepgen -i
ANTECHAMBER_PREP.AC -f int -o sustiva.prepin -rn "SUS " -rf
molecule.res |
最後には表2のファイルが作業ディレクトリに作られる。
表2
|
ANTECHAMBER_AC.AC
ANTECHAMBER_PREP.AC
divcon.rst ANTECHAMBER_AC.AC0
ANTECHAMBER_PREP.AC0
NEWPDB.PDB ANTECHAMBER_AM1BCC.AC
ATOMTYPE.INF PREP.INF ANTECHAMBER_AM1BCC_PRE.AC divcon.dmx
sustiva.prepin ANTECHAMBER_BOND_TYPE.AC divcon.in ANTECHAMBER_BOND_TYPE.AC0 divcon.out |
.
大文字で示すファイルはantechamberが使う中間ファイルであり、以降では必要がないので削除してもよい。うまくいかない場合、チェックすることが出来るので初期設定では削除しないdivcon.xxx files はAntechamber がatomic point charges を計算するために用いるdivcon quantum mechanics code のinputとoutputファイルで ある。ここでは、divcon calculationが完全に実行されたかどうかをチェックする以外には用いない。
表3 divcon.out
|
****************************************************************************** *
* *
DIVCON 99a
* * *
CYCLE = 431
TIME =
0.400 ENERGY = -120.030094
GNORM =
0.537 GRDMAX = 0.1988 GRDAVR = 0.0429 DELTAE =
-0.000057 DELTAX = 0.000029 ENERGY TEST PASSED COORDINATE TEST PASSED GRADIENT NORM TEST PASSED GRADIENT COMPONENT TEST PASSED -- GEOMETRY OPTIMIZED -- FINAL QUANTITIES: ----------------- |
ここで興味あるファイルは、susutiva.prepinで、Antechamberの実行はこのファイルを作成するためである。
表4 sustiva.prepin
|
0 0 2 This is a remark line molecule.res SUS INT
0 CORRECT OMIT DU BEG
0.0000 1
DUMM DU M 0 -1 -2
0.000
.0 .0 .00000 2
DUMM DU M 1 0 -1
1.449
.0 .0 .00000 3 DUMM DU M 2
1 0 1.522 111.1 .0 .00000 4
C2 ca M 3 2 1 1.540 111.208
180.000 -0.17826 5
C1 ca M 4 3 2 1.386 86.897
-162.979 -0.04968 6
H1 ha E 5 4 3 1.072 120.119 25.599 0.16937 7
C14 ca M 5 4 3 1.379 119.724
-154.668 -0.01913 8
Cl1 cl E 7 5 4 1.742 119.676
-179.857 -0.07512 9
C13 ca M 7 5 4 1.385 120.634 0.368 -0.06683
10 H9 ha
E 9 7 5 1.073 120.065
179.750 0.15651
11 C12 ca M
9 7 5 1.380 119.705 0.192 -0.17242
12 H8 ha E 11 9
7
1.076 120.047 179.373 0.14949
13 C11 ca M
11 9 7 1.388 119.964 -0.551 0.10869
14 N1 n M 13 11
9 1.388 121.155 179.940 -0.47390
15 H7 hn E 14 13 11 0.995 120.171 3.766 0.35663
16 C10 c M
14 13 11 1.362 124.462
-166.513 0.84152
17 O2 o
E 16 14 13
1.183 123.299 172.521 -0.58069
18 O1 os M 16 14 13 1.338 115.711 -5.631 -0.37631
19 C3 c3 M 18 16 14 1.416 124.597 -18.703 0.31502
20 C9 c3 3 19 18 16 1.543 105.801 -88.841 0.61999
21 F1 f E 20 19 18 1.321 110.093 61.264 -0.22897
22 F2 f E 20 19 18 1.319 110.789 -179.348 -0.23078
23 F3 f E 20 19 18 1.311 111.555 -58.566 -0.21487
24 C4 c1 M 19 18 16 1.470 107.200 154.846 -0.19720
25 C5 c1 M 24 19 18 1.186 179.487 175.816 0.01368
26 C6 cx M 25 24 19 1.449 179.653 -134.955 -0.07873
27 H2 hc E 26 25 24 1.075 114.014 53.214 0.10860
28 C7 cx M 26 25 24 1.507 119.531 -91.769 -0.10741
29 H3 hc E 28 26 25 1.075 116.902 141.614 0.08023
30 H4 hc E 28 26 25 1.074 117.386 -0.494 0.08282
31 C8 cx M 28 26 25 1.489 60.373 -109.125 -0.11247
32 H5 hc E 31 28 26 1.075 118.643 -106.493 0.07951
33 H6 hc E 31 28 26 1.075 118.172 107.331 0.08074 LOOP
C11 C2 C3
C2 C8
C6 IMPROPER C3 C11 C2
C1 C2
C14 C1 H1 C1
C13 C14 Cl1
C12 C14 C13 H9
C11 C13 C12 H8
C12 C2 C11 N1
C10 C11 N1 H7 N1
O2 C10 O1 DONE STOP |
このファイルには、sustiva残基の全てのチャージ、原子タイプ、が含まれ、それらにより、Leap を使ってprmtopとinpcrdファイルを作ることになる。
このファイルには、sustiva 分子の3D構造、それぞれの原子上のチャージ、原子の番号(column 1)、名前 (column 2) アトムのタイプ (column 3)が含まれている。 loopと不適当な torsionも示されている。しかし、このファイルにはパラメータは含まれていない。GAFF parameters は全て$AMBERHOME/dat/leap/parm/gaff.datに書かれている.。またここで示されているGAFF atom typesは、全て小文字で表されている。これは、GAFF
force field がmacromolecular AMBER force fieldsと独立して扱われるためである。普通、 AMBER
force fields は大文字の原子タイプで示され、GAFF と traditional force fields が同じ計算のなかで混同されないようになっている。
ほとんどのbond,
angle と
dihedral parametersパラメータは、このファイル中で定義されてはいるが、まだ定義されずに残っているものがある可能性もある。その場合、定義されていないパラメータをprmtopとinpcrdファイルをLeapで作成する前に調べておく必要がある。そのために、parmchkユーティリティを用いる。
>parmchk -i sustiva.prepin -f prepi -o sustiva.frcmod
|
remark goes here MASS BOND ANGLE ca-c3-c1 64.784 110.735 Calculated with empirical approach c1-c1-cx 56.400 177.990 same as c1-c1-c3 c1-cx-hc 48.300 109.750 same as c1-c3-hc c1-cx-cx 64.200 111.590 same as c1-c3-c3 DIHE IMPROPER NONBON |
このコマンドを実行し、sustiva.frcmodファイルを作成する。このパラメータファイルは、 Leapを実行する際に定義されてないパラメータを加えるために使用する。antechamber は、missing parametersを 同様のパラメータに対する analogy
で可能な限り作成する。. しかし、シミュレーションの前に、これらのパラメータを少し注意深くチェックする必要がある。Antechamber が経験的にそれらの値を計算することが不可能であったり、アナロジーが無理な場合は、最もリーゾナブルだと思われる初期値を記入するか、ゼロを入れ"ATTN: needs revision"というコメントを書き加えるからである。その場合は自分でそれらのパラメータを加えなければならない。 GAFF が出来るだけパラメータを増やしてくれることを希望したい。表5は作成されたfrcmodである。
表5 sustiva.frcmod
4つのmissing angle parametersが書かれている。Xleapで後ほどどの原子がこれらに相当するかを検討する。このチュートリアルではAntechamber が示したパラメータは正しいと仮定する。本来は例えばab initio
calculations などでこれらのパラメータを検討するのが望ましい。これまでの作業でLeapでsustiva関係した必要なすべての情報をそろえた。Leap を実行し、GAFF force field が使用可能かどうかを検討する。traditional AMBER force fields と GAFF を一緒に使えることが可能なので、ここでa
fragment of the HIV virus を入力しFF99 force fieldで扱い、sustiva 分子をGAFF force fieldで扱う。このチュートリアルでは、GAFF sustivaを読み込み、truncated octahedral box of TIP3P waterに埋め込む。TIP3P water parameters はFF99 Leap scriptの一部として読み込まれるので、Xleapは以下のようにして実行できる(生命環境のコンピュータは、単にxleap enterのみで下記の実行と同じ条件で実行可能)。
|
>xleap -s -f $AMBERHOME/dat/leap/cmd/leaprc.ff99 |
Xleap が開始したら、GAFF force fieldが使えるかどうかを確認する。 $AMBERHOME/dat/leap/cmd/ にスクリプトがあるので、以下のようにしてそれが確認可能である。
>source leaprc.gaff
OurXLeap window に以下のように表示される。
そこでsustiva.prepinを入力する
>loadamberprep sustiva.prepin
listと入力すると、SUSという新しいunitを確認することが出来る。このunitはprepinファイルの5行目にある名前と同じ名前である。
Pramchkで作成したfremodファイルを入力してないのでSUS をチェックすると以下のように表示され、4つの4 missing angle type parameters が有ることを確認できる。
check SUS
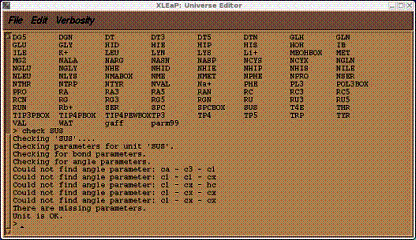
Sustiva unitのどの原子にそれらが相当するかを見るために、以下のコマンドを入力する。
>edit
SUS
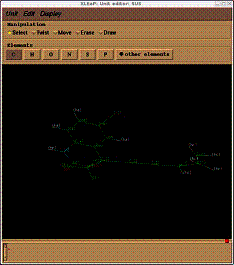 Display->Types
Display->Types
missing angle type parameters が ca-c3-c1, c1-c1-cx, c1-cx-hc and c1-cx-cxに相当することが分かる。これらはpropyl
ring と c-c
triple bondである。これらは有機化合物であまり無いことからも予測されることである。このウィンドを閉じ、frcmodファイルを入力しXleapにmissing angle typesのデータを与える。
.
>loadamberparams sustiva.frcmod
SUS unitをチェックすると、missing parametersが無いことを確認できる。そこで、
水を加えprmtop とinpcrd fileを作成する。
>solvateoct SUS TIP3PBOX 10
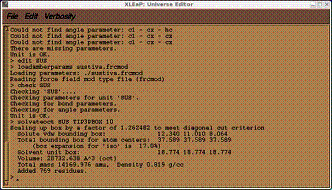
これで10オングストロームのエッジをもつsustiva
molecule の全ての原子から最少距離にあるpre-equilibrated TIP3P water のtruncated octahedral box を作成することが出来る。Edit SUS
と入力し確認する。
>edit SUS
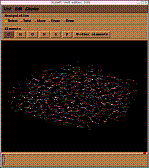
これでtopology とcoordinate filesファイルを以下の様に入力し作成する。
>saveamberparm SUS sustiva.prmtop sustiva.inpcrd
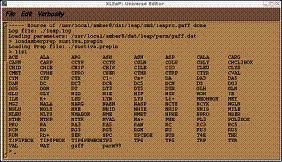
これで必要なファイルが作成され、
シミュレーションを開始することが可能である
1
Wang, J., Wolf, R.M., Caldwell, J.W., Kollman, P.A., Case, D.A.
"Development and Testing of a General Amber Force Field", J. Comp.
Chem., 2004, 25, 1157 - 1173.
2 Jakalian, A., Bush, B.L., Jack, B.D., Bayly, C.I., "Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method.", J. Comp. Chem., 2000, 21, 132-146.